
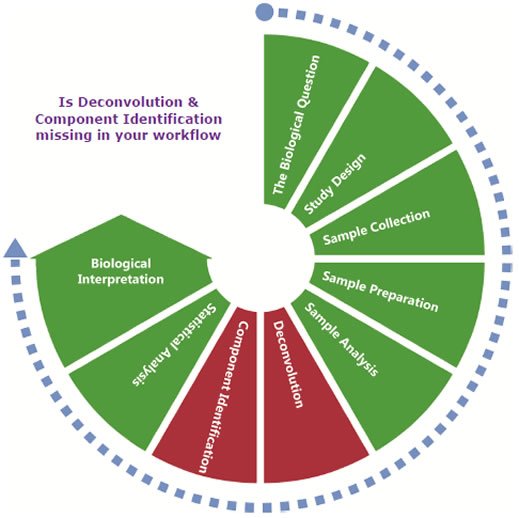
Using AnalyzerPro® to Complete your Metabolomics Workflow
First Published: Metabolomics Society Conference, SECC Glasgow 2013
A Case Study from Murdoch University
Murdoch University is one of the five nodes of Metabolomics Australia. They have developed a successful and respected workflow for their GC-MS metabolomics studies which is presented here.
The Biological Question
The most important part of any metabolomics study is the specific biological question that the study is designed to answer. This question should be used to define the overall study and should be objective, measurable, realistic and relevant. For example:
Is there a difference in the secreted metabolome of Salmonella grown as a biofilm or in a planktonic form?
or:
Which metabolites are up-regulated in the bloodstream following inflammation?
Without this biological question it may be impossible to make sense of the data or to design the study appropriately. It is not sufficient to simply collect as much data as possible and then attempt to explain observations.
Some things in life we can do without, in Metabolomics you can’t do without AnalyzerPro. Reliable, Powerful and Intuitive software.
Study Design
It may seem obvious, but the study should be designed to answer the biological question. As we are dealing with biological processes it is critical that we have enough samples in the study to account for natural biological variation. At Murdoch University, the preference is for 8 biological replicates per treatment or control. The Metabolomics Society recommends 5 replicates and the minimum number required for publication is three. The actual number of samples required for a given study will vary depending on a number of factors. For example, human studies can be particularly problematic due to the difficulty in standardising human subjects and as such require the greatest number of samples.
Sample Collection
Optimal sample collection methods are not always achievable, nevertheless samples must be collected in a consistent manner. If the samples contain cells which are still metabolically active, this metabolism must be quenched usually by snap-freezing or with cold solvents. Samples should be frozen and kept at as low a temperature as possible. However there is no consensus as to how long samples remain viable for metabolomics studies even when frozen at -80°C. In general, metabolomics studies are more time sensitive than the other ‘omics’ (Proteomics and Genomics).
Sample Preparation
The sample preparation required will vary depending on the analytical technique(s) chosen. Most methods require an extraction step to remove proteins and, if necessary, break open intact cells. The extraction stage can also be used to concentrate the analytes of interest and introduce a suitable internal standard. Analysis by GC-MS requires that the analytes are volatile and this may require the preparation of suitable derivatives. Sample preparation should also consider the requirements for long term sample storage, not forgetting that samples are usually best kept as frozen dried extracts.
Sample Analysis
At Murdoch, most of the instrumentation is fully automated, requiring minimal intervention from the user. In other laboratories, people use RemoteAnalyzer® to simplify and unify the interface for submitting samples for acquisition particularly when multiple instrument vendors’ instruments are available.
Replicate sample analysis is required to take account of natural variation. The biological variation will usually be considerably greater than the analytical variation so biological replicates are preferred over analytical replicates. It is important to consider how long samples are likely to remain viable as well as how frequently the analytical instrument requires maintenance and calibration when planning the frequency and positioning of replicates.
Depending on the design of the Metabolomic study, the MS data may be acquired in Full Scan, SIM or MRM mode. Data from multiple vendors can be imported into AnalyzerPro for data processing.
Deconvolution
Data from the analytical instrument needs to be processed before it can be analysed and interpreted.
Our first step in data processing is called ‘deconvolution’ which is [helpfully] defined as ‘reversing the effects of convolution’. For chromatographic data the biggest problem is co-elution of chromatographic peaks. Many components will not be completely separated by the GC column. Using AnalyzerPro we can determine which masses belong to which of the co-eluting peaks. The deconvolution step also allows the creation of target component lists. For comprehensive data analysis automated library building is available in AnalyzerPro which includes all the components within an entire data set. This differs from a traditional target component list generated from standards in that the components do not require to be identified until later in the analysis. At this stage, it is only important that they are there. The list produced from the automated library building stage will be comprehensive and the component list will be very large. As such, there is a fundamental need for a detailed level of quality control. The upside of this is that your data set can be completely defined by the component list generated during the automated library building stage.
Component Identification
Following deconvolution, each component found can be assigned an identity. This identity does not have to be absolute and subsequent analysis can be performed on identified components or by treating each component as an unknown. Components can be identified from libraries or from a specific target component library. A target component library is usually based on standards which have been run on in house instruments as this provides more reliable data. Absolute identity of all components is not required at this stage but where a component of interest is found (i.e. where a difference is observed) that component should be investigated further to confirm its identity.
Statistical Analysis
The most common way to make sense of large metabolomics data sets is by the use of multivariate statistical analysis. Principal Component Analysis (PCA) allows the data to be modelled and to determine which metabolites [components] are contributing most to the variance that has been observed. It also allow the identification of outliers that are likely to contribute to the skewing of the variance of a particular component. PCA points you in the direction needed for interpretation, but is only one part of the data analysis flow.
The next step is to scrutinise those components contributing most to the variance. It is important to differentiate between components that may be derivatisation artefacts or interferences which are of no biological interest (which can legitimately be removed or their variance accounted for) and those that are identified as causing variation in the data sets that require further investigation.
Biological Interpretation
At this point in the workflow, can we answer the biological question that was posed at the beginning of the study or is there now a new question? In either case we have moved forward in our understanding, or lack of, following this Metabolomics workflow. Subsequent data analysis can involve the mapping of metabolites to pathways and looking at the up or down regulation of specific metabolites. However, we may have indeed found that we have answered our original question, additionally though, we may have also generated many other questions. In this situation, the process of metabolomics research begins again, starting with a new biological question and if necessary, revisiting the original data set. These stages are customary practice in the analysis of Metabolomic data sets.
Conclusion
AnalyzerPro is a key element in this Metabolomics workflow. The targeted and non-targeted approach of the peak picking and deconvolution functionality is applicable to large data sets from multiple vendors’ instruments. The deconvolution of complex chromatographic separations is equally applicable to GC-MS and LC-MS analyses.
Acknowledgements
Robert Trengove, Garth Maker, Catherine Rawlinson & Joel Gummer – Murdoch University, Perth Australia.