
Scott J Cambpell1, SJ Roberts1, J Wingate2, B Kieser2, T Sakuma3, P Vollmerhaus3
1SpectralWorks Ltd, The Heath Business & Technical Park, Runcorn, UK; 2Applied Biosystems Inc, Concord, ON, Canada; 3MDS Sciex, Concord, ON, Canada
First Published: PittCon 2004
Introduction
Traditionally high resolution mass spectrometers have been expensive and complicated to use. The primary uses for high resolution mass spectrometry were for molecular mass confirmation and elemental composition elucidation of a single compound. However, the recent advent of a generation of high resolution mass spectrometers that are affordable, relatively easy to use and can be operated on a chromatographic time scale have broadened the application base for the technology. Modern mass spectrometers can provide accurate molecular mass and elemental composition analysis of complex mixtures, as well as low detection and quantitation limits for the analytes. Nonetheless, much of the value of the data may be lost as traditional data mining packages treat the data as if it is nominal or at best low precision. In this poster we illustrate, through the use of novel data processing algorithms, that it is possible to process complex samples and maintain the integrity of high resolution accurate mass data from the HPLC/MS analysis of a number of amino acids.
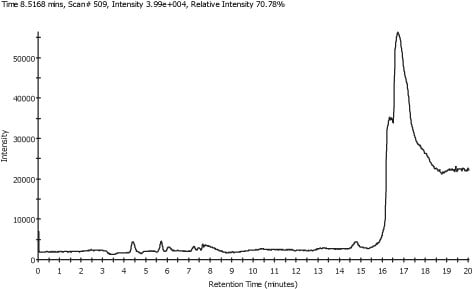
Figure 1. Typical Plasma TIC
The use of high resolution, accurate mass, mass spectrometers can allow users to identify both known and unknown components. This can be done with a greater amount of certainty than if a unit mass resolution instrument were used. A problem occurs however when non targeted analysis are under-taken as most commercially available software packages are not capable of taking advantage of the mass accuracy of the data when searching for unknown components within a complex sample. Software applications that can handle accurate mass tend to process the data very slowly. In this example, the analysis of samples of plasma spiked with amino acids and analyzed by high resolution mass spectrometry is presented.
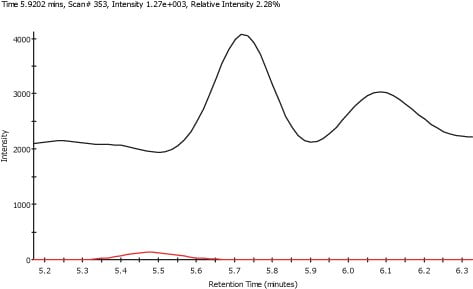
Figure 2. XIC for Serine (lower) and TIC (upper)
Materials and Methods
Samples of plasma were spiked with known amounts of 19 amino acids. HPLC/MS analysis were carried out under the following conditions:
| Column: | Waters 100mm x 4.6mm |
| Spherisorb S5CN | |
| Solvent A: | Water, 10 mM Ammonium Formate |
| 0.1% Formic Acid | |
| Solvent B: | Acetonitrile, 10 mM Ammonium |
| Formate 0.1% Formic Acid | |
| Gradient: | 100% A for 5mins to 100%B in |
| 11mins held for 15mins | |
| MS: | ABI/MDS Sciex QSTAR ® |
| Ionization: | TurboIonSpray ® Ion Source |
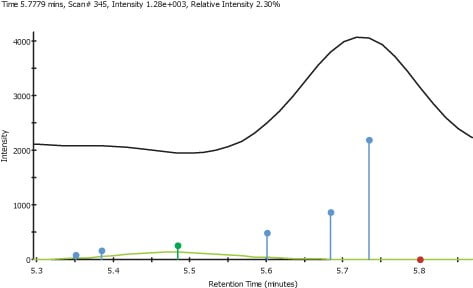
Figure 3. Found, Identified and not found components
Figure 1 shows a typical Total Ion Chromatogram (TIC) obtained from a plasma sample. As is common with LC/MS data, the TIC is complex and does not clearly show the peaks from the amino acids that have been spiked into the samples. However, by using extracted ion chromatograms (XIC) it is possible to produce chromatograms for each of the amino acids. Figure 2 shows the XIC for Serine, mass 122.03, and the TIC.
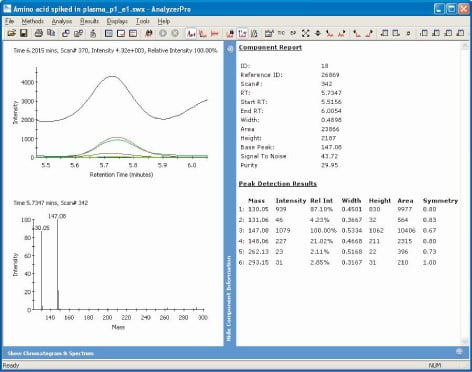
Figure 4. AnalyzerPro graphical report
The use of XIC is useful for finding known components, although it can become time consuming if either the number of components or the number of analyses becomes large. The use of manually preparing large numbers of XIC becomes impractical when we need to exhaustively search for unknown or non-target components. An alternative approach is to use the software package Analyz-erPro. AnalyzerPro is able to analyze complex samples automatically and rapidly extract com-ponents at accurate mass from the TIC. The system can also apply target component analysis to confirm or refute the presence of specific components. A total of 81 components were observed in a single plasma sample. Examples of found (non-target), identified (target) and not found target components are shown in Figure 3. It shows a number of found components, a tar-geted component that was found at RT 5.48 (Component ID 15) and a component that was targeted but not found at RT 5.8, as indicated with the blue, green and red dots respectively.
In Figure 4 we can see how AnalyzerPro graphically displays the spectrum for the found component and each of its ions as XIC, as well as a textual report of the component.
Conclusion
A high resolution accurate mass HPLC-MS dataset was successfully analyzed for both target and non target components using AnalyzerPro. This allowed the reliable, rapid and automated picking of chromatographic components within a com-plex plasma sample matrix . The amino acids that were spiked into the plasma were identified by use of their accurate mass and chromatographic retention time. Additional non target components were also observed and accurate mass spectra obtained without compromising data processing time. The data analysis capability of AnalyzerPro allows accurate masses to be found from raw data thus aiding the identification of both known and unknown components. This is particularly useful when dealing with LC/MS data as traditionally the TIC can obscure sample components.